
Synthetic Lethality (SL) is a type of genetic interaction in which the simultaneous inactivation of two genes leads to cell death, while the inactivation of one gene alone does not affect the viability of the cell. In a pair of SL genes, if one gene has a cancer-specific mutation, the other gene can be a potential drug target because perturbation of the latter gene can selectively kill cancer cells while sparing normal cells. SL interactions are identified mainly by experimental screening and computational prediction, but experimental screening is costly and time-consuming. Thanks to the accumulation of SL data, data-driven machine learning models for SL prediction have been actively developed in recent years. However, most recent machine learning methods learn the representation of each gene individually, ignoring the representation of pairwise interactions between two genes. In addition, the mechanism of SL, which is the key to translating SL into cancer therapeutics, remains unclear.
SIST Associate Professor Zheng Jie's group proposed a graph neural network named PiLSL based on pairwise interaction learning, to learn the representation of the pairwise interaction between two genes from a knowledge graph constructed by the group—SynLethKG for SL prediction. In addition, PiLSL utilizes the attention mechanism to explain the SL mechanism through weighted paths in the enclosing graphs. Extensive experimental results show that PiLSL has superior performance over the state-of-the-art baseline methods and has strong generalization ability in three realistic settings.
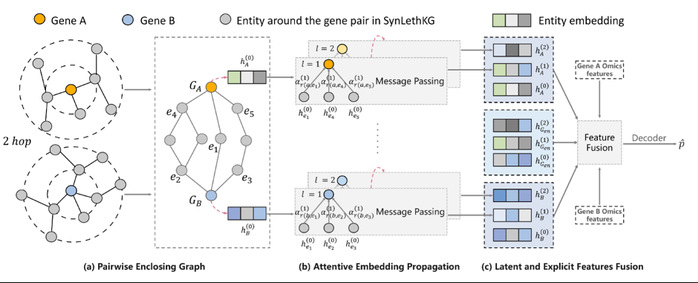
Overview of PiLSL. (a) Extracting an enclosing graph for each gene pair; (b) Learning gene representations from the enclosing graph using the graph attention mechanism; (c) Fusing the implicit features learned from the enclosing graph and the explicit features learned from multi-omics data to obtain the representation of the gene pair
Apart from the shortcomings in gene representation, another limitation of the supervised machine learning methods is that they usually formulate SL prediction as a task of binary classification, training predictive models against known SLs as positive samples and some other gene pairs as negative samples. However, it is difficult to obtain high-quality data of negative samples, which is a bottleneck of SL prediction methods based on the classification task. Besides, in practice, considering the high cost of wet-lab experimental validation, users are often more concerned about the percentage of true positives in the top list of predicted SL pairs, rather than the overall performance of classification in the whole list of thousands of candidates.
To avoid the dependence on high-quality negative samples data, a contrastive learning-based model named NSF4SL for SL partner gene recommendation is proposed. NSF4SL formulates SL prediction as a gene ranking problem, and relies on the positive SL data to learn the similarity among SL gene pairs. In addition, NSF4SL designs a data augmentation method to incorporate global statistics of the training data into the gene representation to address the issue of SL data sparsity. Experimental results show that NSF4SL outperforms all the baseline models which require negative samples, and has better generalization ability in various experimental settings.
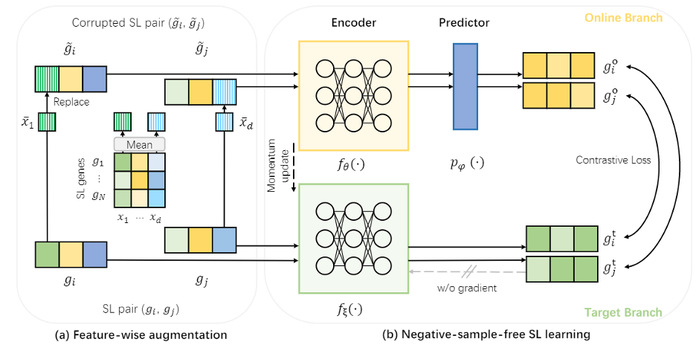
Overview of NSF4SL. (a) Feature-wise augmentation for input gene features; (b) Negative-sample-free SL learning based on the contrastive learning framework
The above research results of Zheng Jie’s group were published in two papers, entitled PiLSL: pairwise interaction learning-based graph neural network for synthetic lethality prediction in human cancers, and NSF4SL: negative-sample-free contrastive learning for ranking synthetic lethal partner genes in human cancers, which were presented (oral presentations) at the 21st European Conference on Computational Biology (ECCB 2022), a flagship conference in Computational Biology.
Second-year master student Liu Xin is the first author of the first paper, and third-year master student Wang Shike is the first author of the second paper. Prof. Zheng Jie is the corresponding author of the two papers.

Wang Shike (left) and Liu Xin (right) at the international flagship conference in Bioinformatics and Computational Biology (ECCB2022)

Liu Xin gave an oral representation on PiLSL at ECCB2022

Wang Shike gave an oral representation on NSF4SL at ECCB2022
PiLSL and NSF4SL are expected to have contributions to innovations in the field of AI-aided drug target discovery. In recent years, Zheng Jie's group has made a series of innovative achievements in the field of machine learning for SL prediction, being one of the leaders in this area. In the future, they will focus on the development of interpretable SL prediction models that integrate multi-omics data, to guide the design of wet-lab experiments for drug target discovery and validation.
**The news article is provided by Zheng Jie

 沪公网安备 31011502006855号
沪公网安备 31011502006855号


